A Distinct, Quiescence-Associated Pluripotent State that is Regulated by Runx1 and can be Modeled In Vitro
Lilyana Margaretha,1,2,3, Brigham H. Mecham4, Christopher Cavanaugh1,7, Daniel C. Jones5, Angelique Nelson1, Jay Shendure6, Walter L. Ruzzo5,6,8, Carol B. Ware1,7, and Carl Anthony Blau1,2,6,*
1Institute for Stem Cell and Regenerative Medicine, University of Washington, Seattle, WA, 98195
2Department of Medicine/Hematology, University of Washington, Seattle, WA 98195
3Graduate Program in Molecular and Cellular Biology, University of Washington, Seattle, WA 98195
4Sage Bionetworks, Seattle, WA, 98109
5Department of Computer Science and Engineering, University of Washington, Seattle, WA 98195
6Department of Genome Sciences, University of Washington, Seattle, WA 98195
7Department of Comparative Medicine, University of Washington, Seattle, WA 98195
8Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA
*To whom correspondence should be addressed.
Submitted.
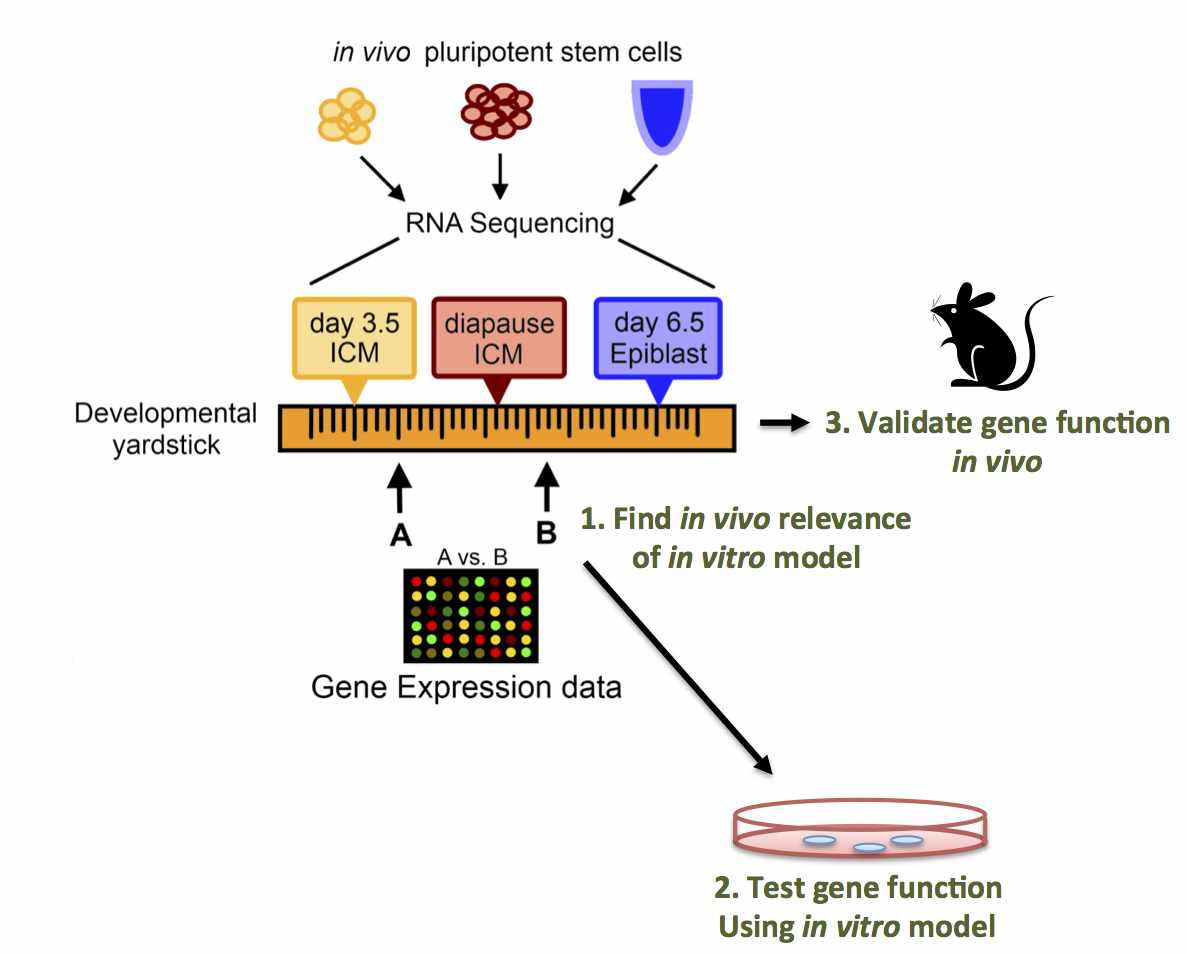 Abstract: Pluripotent stem cells inhabit, and can transition
between, distinct metastable states. Embryonic stem cells (ESC)
derived from the pre-implantation inner cell mass (ICM) can be
maintained in a ground state of pluripotency, whereas epiblast stem
cells (EpiSC) from the post- implantation embryo are primed for
differentiation. We hypothesized that a physiological state of
dormancy known as diapause represents a novel pluripotent state. We
performed RNA-Sequencing on pluripotent stem cells isolated from: i)
the ICM of day 3.5 pre-implantation embryos; ii) the epiblast of day
6.5 post-implantation embryos; and iii) the ICM of day 8.5 embryos
induced to enter diapause. Our results define diapause as a distinct
pluripotent state and enable the construction of a "developmental
yardstick" against which cultured pluripotent stem cells can be
compared. Using this yardstick, we describe culture conditions that
mimic diapause, and identify Runx1 as a regulator of this "quiescence-
associated" state.
Abstract: Pluripotent stem cells inhabit, and can transition
between, distinct metastable states. Embryonic stem cells (ESC)
derived from the pre-implantation inner cell mass (ICM) can be
maintained in a ground state of pluripotency, whereas epiblast stem
cells (EpiSC) from the post- implantation embryo are primed for
differentiation. We hypothesized that a physiological state of
dormancy known as diapause represents a novel pluripotent state. We
performed RNA-Sequencing on pluripotent stem cells isolated from: i)
the ICM of day 3.5 pre-implantation embryos; ii) the epiblast of day
6.5 post-implantation embryos; and iii) the ICM of day 8.5 embryos
induced to enter diapause. Our results define diapause as a distinct
pluripotent state and enable the construction of a "developmental
yardstick" against which cultured pluripotent stem cells can be
compared. Using this yardstick, we describe culture conditions that
mimic diapause, and identify Runx1 as a regulator of this "quiescence-
associated" state.
Supplementary RNA-seq Information:
This web site provides supplementary information about our RNA-seq experiments, including processed RNA-seq results in formats suitable for viewing as "Custom Tracks" in the UCSC Genome Browser.
RNA-Seq Library Preparation
Total RNA was extracted using QIAGEN Micro RNeasy kit and amplified following protocols of WT-Ovation Pico kit (Nugen). The quality and quantity of RNAs were determined on an Agilent 2100 Bioanalyzer (Agilent Technologies). cDNA amplification products were made into double-stranded cDNA using WT-Ovation Exon Module kit (Nugen). 3 ug of cDNA product was sheared by sonication (Bioruptor UCD-200) for 2 x 15 min at 4oC, H speed (320 W). cDNA fragments were then blunt-ended with End-It DNA End-Repair kit (Epicentre Biotechnologies) and A-tail was added to the end of the fragments. Adapters (33-34 nucleotides) were ligated to each end. The fragments were subjected to size selection by gel electrophoresis to isolate 200-300 bp fragments, which were further enriched by 18 cycles of PCR amplification. Cluster generation of the cDNA library was performed on a cBot Cluster Station (Illumina, Inc.) and the samples were sequenced on Genome Analyzer IIx (Illumina, Inc.).
RNA-Seq Read Alignment and Analysis
Reads were aligned using Bowtie (Langmead et al., 2009) [2] version 0.12.5, to version mm9 of the mouse genome, obtained from the UCSC genome browser (Fujita et al., 2010) [1]. Paramaters were adjusted to allow for two mismatches in the initial eighteen bases. To account for reads crossing splice junctions, we aligned the remaining reads to a set of sequences taken from upstream of the donor site and downstream of the acceptor site of every splice junction annotated in version 58 of the Ensembl gene annotations (Hubbard et al., 2009) [4]. For the purpose of analysis we used only reads aligned uniquely to the genome, and took the additional precaution of filtering out any reads overlapping low complexity regions or simple repeats as defined by the UCSC genome brower RepeatMasker track (Smit et al., 1996-2010; http://www.repeatmasker.org) [3]. The browser tracks provided on this page also exclude these low complexity reads. Reads overlapping other classes of repeats, in particular commonly transcribed elements such as LTR, rRNA, tRNA, scRNA, etc. are retained. After summarizing the expression of each gene by the number of reads aligned unambiguously within any of its exons, the DESeq package was used to perform differential expression analysis (Anders and Huber, 2010) [5].
Browser Tracks
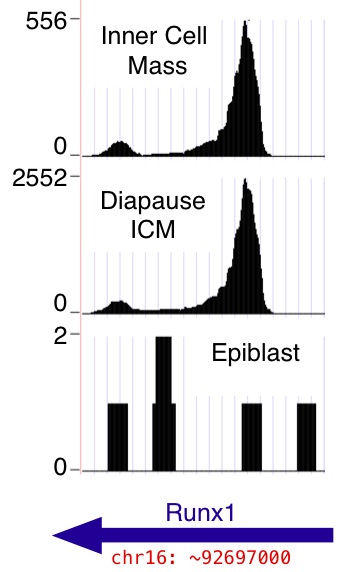 Each data set is a browser "wiggle" track, provided in
.bigWig
format. Download these files if desired (use the "Download" links).
Additionally, they may be viewed in the browser without downloading
them to your computer. (Use the "View in Browser" links. Return
and click additional ones to see several at once, or use the "View
All" link; the browser retains your preferences.)
Each data set is a browser "wiggle" track, provided in
.bigWig
format. Download these files if desired (use the "Download" links).
Additionally, they may be viewed in the browser without downloading
them to your computer. (Use the "View in Browser" links. Return
and click additional ones to see several at once, or use the "View
All" link; the browser retains your preferences.)
In all cases, the y-axis in the "wiggle" track is the number of mapped 36 base reads that include the given nucleotide position in the mm9 genome assembly. Reads from both genomic strands are combined. Note that the number of mapped reads differs from sample to sample (see Table 1 in the main paper; totals are reproduced below for convenience). The per-nucleotide read counts shown in the wiggle tracks are absolute counts, not normalized to correct for variation in per sample read depth. Also note that by default the browser dynamically rescales the y axes so that the highest visible peak in each track occupies the full track height ("auto-scale to data view"); scales appear near the left edge of the browser window. See WiggleTrackHelp for help with browser wiggle track viewing options.)
| Sample | Mapped Reads | View | Download | ||||
|---|---|---|---|---|---|---|---|
| Inner Cell Mass, replicate 1 | 2,844,162 | View in Browser | Download: | ICM1.bigWig ( 9M) | |||
| Inner Cell Mass, replicate 2 | 3,023,359 | View in Browser | Download: | ICM2.bigWig ( 8M) | |||
| Inner Cell Mass, replicate 3 | 7,979,055 | View in Browser | Download: | ICM3.bigWig (22M) | |||
| diapause Inner Cell Mass, replicate 1 | 3,743,553 | View in Browser | Download: | dICM1.bigWig (10M) | |||
| diapause Inner Cell Mass, replicate 2 | 3,572,446 | View in Browser | Download: | dICM2.bigWig ( 9M) | |||
| diapause Inner Cell Mass, replicate 3 | 7,581,714 | View in Browser | Download: | dICM3.bigWig (20M) | |||
| epiblast, replicate 1 | 2,173,025 | View in Browser | Download: | epi1.bigWig ( 7M) | |||
| epiblast, replicate 2 | 2,177,578 | View in Browser | Download: | epi2.bigWig ( 8M) | |||
| View All | |||||||
References
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2010 Oct 18.
- Langmead, B., Trapnell, C., Pop, M., & Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 10(3), R25. doi:10.1186/gb-2009-10-3-r25
- Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. http://www.repeatmasker.org. 1996-2010.
- Hubbard, T. J. P., Aken, B. L., Ayling, S., Ballester, B., Beal, K., Bragin, E., Brent, S., et al. (2009). Ensembl 2009. Nucleic acids research, 37(Database issue), D690-7. doi:10.1093/nar/gkn828
- Anders, S., & Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biology, 11(10), R106. BioMed Central Ltd. doi:10.1186/gb-2010-11-10-r106
If you have problems with this web page, please contact ruzzo![]() uw.edu.
uw.edu.